原发皮肤间变大细胞淋巴瘤
Primary Cutaneous Anaplastic Large Cell Lymphoma, C-ALCL
发布时间:2016-04-07 21:49:38
概述:
诊断要点:
1. 中位发病年龄60岁(儿童偶有报道),主要累及躯干、面部、和四肢。表现为孤立性结节,也可多发(20%病例),常伴溃疡。偶尔病灶自行消失,但复发。少数病例(10%)可以扩散至皮外部位,主要是局部淋巴结。
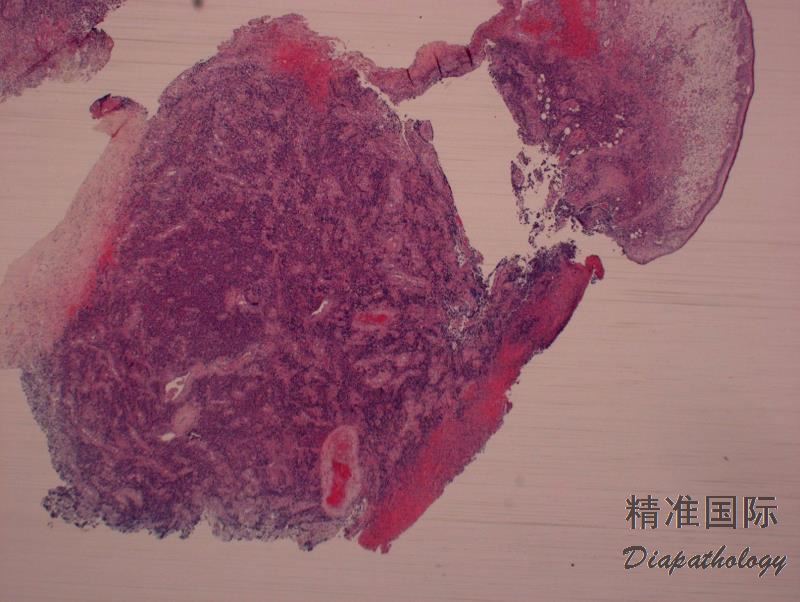
2. 肿瘤细胞弥漫浸润,主要累及真皮层,可延及皮下组织,但有时见亲表皮浸润,特别是有DUSP22-IRF4病例。病灶周围可见少量背景炎性细胞,但如有溃疡,则常伴有丰富炎性细胞常(类似LyP)。罕见病例含大量中性粒细胞(中性粒细胞富有亚型)。
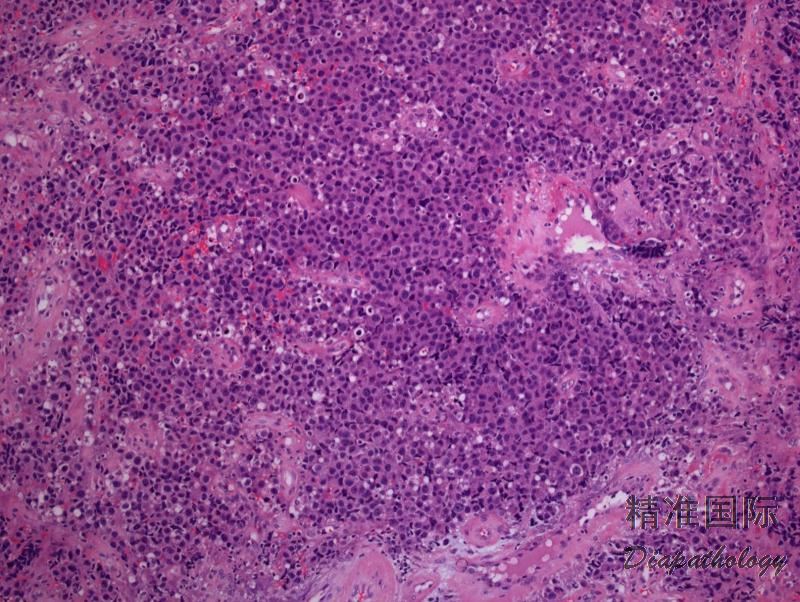
3. 多数病例肿瘤细胞呈现间变性形态(园或椭圆形核、核不规则、显著嗜伊红核仁、丰富胞质),但也可呈现多形性或免疫母样细胞形态。
4. 免疫表型:肿瘤细胞表达CD30(>75%的细胞)和细胞毒分子,常表达CD4,但其它T细胞相关抗原不定;偶见CD8+/CD4-或CD4-/CD8-,或无任何T相关抗原表达。EMA和ALK一般阴性。约40%病例CD15+,MUM1几乎总是阳性。PAX5阴性,EBER阴性。
5. 大多数病例有TCR基因克隆性重排。绝大多数病例无ALK 基因易位。有极少数报道有t(2;5)或免疫组化ALK阳性(核或核+胞浆),这些病例预后好。25%病例(以及很少数LyP病例)有DUSP22-IRF4易位。

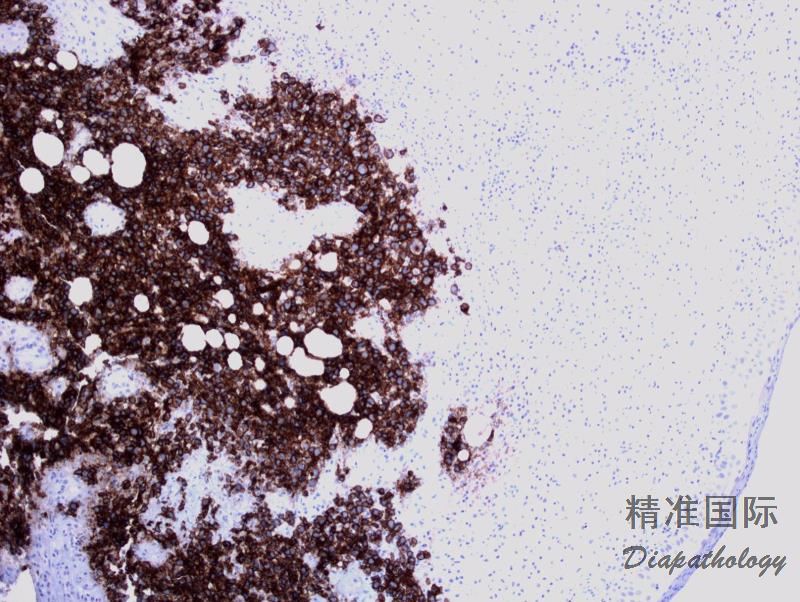


免疫组织化学染色:
鉴别诊断:
2.淋巴瘤样丘疹病(LyP):C 型LyP 形态学和免疫表型可与C-ALCL重叠。同时存在不同发展阶段的皮肤病变及某一病变常在3-12周自行消失支持LyP。
3.蕈样霉菌病(MF)大细胞转化:转化大细胞可以CD30+, 与C-ALCL难以鉴别。MF病史或同时存在不同阶段的典型MF皮肤病变(斑片、斑块)或组织学见到残存的典型MF 细胞及亲表皮浸润,有助诊断。
4.原发性皮肤γ/δT 细胞淋巴瘤(Primary cutaneous γ/δT cell lymphoma):临床表现为侵袭性,肿瘤主要侵犯四肢可也累及全身皮肤和内脏。肿瘤细胞可浸润表皮、真皮或皮下,表达CD3、CD56、CD2、TCRδ,不表达CD4、CD8(少数+)、CD5、βF1。
5.外周T细胞淋巴瘤,非特指(PTCL,NOS):累及皮肤时,表现为斑块或肿瘤结节。组织学表现肿瘤细胞主要弥漫或结节性侵犯真皮,形态大小不定,CD30阴性或局灶阳性,患者有系统性/结性T细胞淋巴瘤病史。
预后:
10年存活期达90%,多灶性或局部淋巴累及不影响预后。